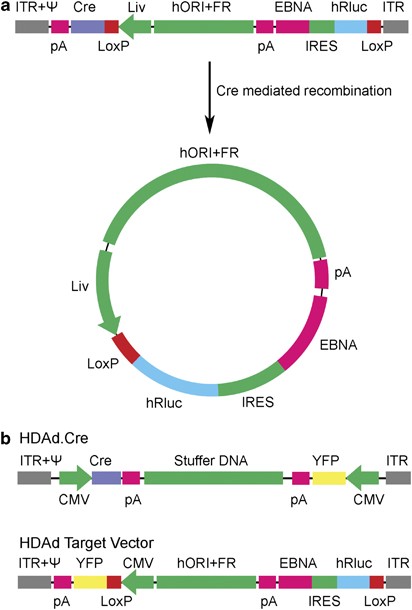
Abstract
Five overlapping Epstein-Barr virus (EBV) recombinants type 1 DNA fragments constituting a complete replication- and transformation-competent genome was cloned into cosmids and transfected together into P3HR-1 cells, along with a plasmid encoding the immediate early activator Z of EBV replication. P3HR-1 cells harbour an EBV type 2 that cannot transform primary B cells due to a deletion of DNA encoding EBNA LP and EBNA 2, but EBV P3HR-1 can provide replication functions in trans and can recombine with the transfected cosmids. EBV recombinants having the EBNA LP type 1 and 2 genes from the transfected EcoRI-A cosmid DNA were selectively and clonally recovered by exploiting the unique ability of the recombinants to transform primary B lymphocytes into lymphoblastoid cell lines.
PCR and immunoblot analyses for seven distinctive markers of type 1 transfected DNAs identified cell lines infected with EBV recombinants that had incorporated EBV DNA fragments beyond the EcoRI-A fragment rescuing the transformation marker. Approximately 10% of the transformant virus recombinants had marker maps at 7, 46 to 52, 93 to 100, 108 to 110, 122 and 152 kbp of the 172 kbp transfected genome. These recombinants likely result from recombination between cloned EBV DNA fragments with transfected cosmids. The only recombinant virus examined in detail by Southern blot analysis has all the polymorphisms characteristic of transfected cosmid type 1 DNA and none characteristic of EBV type 2 P3HR-1 DNA.

This recombinant was wild-type in primary B cell infection, growth transformation, and lytic replication. Overall, the EBNA 3A type 1 gene was incorporated into 26% of the rescued recombinants with transformation markers, a frequency that was considerably higher than that observed in previous experiments with cotransfections of EBV DNA from two cosmids into P3HR cells. -1 (B. Tomkinson and E. Kieff, J. Virol. 66:780-789, 1992). Of the recombinants that had incorporated the cosmid DNA fragment rescuing the marker and the fragment encoding the EBNA 3A type 1 gene, most had incorporated markers from at least two other transfected cosmid DNA fragments, indicating a propensity for multiple homologous recombinations.
The frequency of incorporation of the unselected transfected type 1 EBNA 3C gene, which is near the end of two of the transfected cosmids, was 26% overall, versus 3% in previous experiments using two-cosmid transfections of DNA from VBS. In contrast, the frequency of incorporation of a 12 kb EBV DNA deletion that was near the end of two of the transfected cosmids was only 13%.
Materials And Methods
- Production of cells and viruses.
Akata, a human Burkitt lymphoma-derived cell line carrying EBV episomes and an EBV-negative Akata cell line were grown in RPMI 1640 medium (Sigma-Aldrich Fine Chemicals, St. Louis, Mo.) supplemented with 10% fetal bovine serum. Virus production from Akata cells was induced by surface IgG cross-linking using rabbit anti-human IgG (DakoCytomation, Carpinteria, CA) as previously described. EBV-negative Akata cells were infected with diluted culture supernatant (1:5 and 1:10), and the infected cells were isolated by selection with G418 (Sigma) (700 µg/ml).
- Southern blot analysis.
Genomic DNAs were extracted by the standard proteinase K-sodium dodecyl sulfate method, followed by phenol-chloroform extraction and ethanol precipitation.
- Plasmids
A BamHI fragment from pUC4K (Pharmacia) containing a kanamycin resistance marker gene was cloned into the BamHI site of pBS246 (Life Technologies) to produce pBS246-km. A SalI (broken)-BamHI fragment of pSG5 (Stratagene) was cloned between Asia (broken) and BamHI sites of pEGFPN1 (Clontech) to produce p(SG5)-EGFP. The EcoRI-NotI fragment of p(SG5)-EGFP containing the EGFP gene was replaced with a synthetic oligonucleotide (5′-AATTCTATGACTCTCTTAAGGTAGCCAAAAGC-3′) containing an I-PpoI recognition sequence to produce p(SG5)-IPpoI. pBS246-km, p(SG5)-EGFP and p(SG5)-IPpoI were used as PCR templates to produce linear targeting constructs for getting recombination.
pGEM9Zf(-)-IPpoI has a unique I-PpoI site that replaces the T7 promoter of pGEM9Zf(-) and is supplied as an I-PpoI enzyme control vector (Promega). The synthetic I-PpoI site (described above) was cloned into pGEM9Zf(-)-IPpoI digested with EcoRI-NotI to construct a double I-PpoI vector with two I-PpoI sites. A BamHI (rounded) fragment of APR-Muc1 (kindly provided by Keiichi Kontani, Shiga University of Medical Sciences, Otsu, Japan, and Toshio Kudo, Tohoku University, Sendai, Japan) containing Muc-1 cDNA was cloned into the HincII site of a double I-PpoI vector to construct pIPpoI-Muc1.
- Electrotransformation of E. coli.
Electrocompetent E. coli DH10B bacterial cells were prepared. Electrotransformation was performed using a Bio-Rad Gene Pulser II electroporation system (0.1 cm cuvette, 1.25 kV, 25 μF, 100 Ω, 40 μl cells)